You can add charge, mass number, and radical to an atom in Marvin JS; furthermore, you can define an alias for any atom as well. All of these properties can be set in the Atom properties dialog.
Currently, you can draw reactions in Marvin JS using simple (straight) reaction arrows, or you can use resonance (two-headed) arrows to display the mesomeric structures of a compound.
The "+" signs between the reactant or product molecules have to be added manually in each case using the Reaction Operator
button on the Tools toolbar. Alternatively, if you select the reactant or the product side, and then press the Reaction Operator button,
the "+" signs will be added automatically between each reactant or product molecule. However, it works only if the selection does not contain other "+" signs or a reaction arrow.
If the structure on the canvas contains only a single step reaction, then this structure will be automatically exported as a "reaction file" by Marvin JS.
This means that in the exported file there will be separate blocks for reactants, products and agents making it possible to use the exported file in a reaction search.
In this case, only those file formats are available in the Save dialog which support reactions (MRV, RXN, SMILES, CXSMILES, SMARTS, CXSMARTS, CML,
RXN V3000, SDF, CSSDF, CDX, SKC). Please note that the conversion of the structure to a reaction file happens on server side and requires the reaction converter webservice to be available.
In the case of multistep reactions (or when the reaction converter webservice is not available), the structure will not be converted to a reaction file.
In this case, there will not be reactant/product/agent blocks in the exported files and the "+" signs and arrows of the reaction(s) will be exported as graphical objects.
Marvin JS supports the detailed display of recation mechanisms by using electron flow arrows (or mechanism arrows) to describe the formation and breaking of chemical bonds. You can use single and double electron flow arrows for representing each elementary step in a complex mechanism. You can read a detalied description about using electron flow arrows in Marvin JS.
Marvin JS provides the option to set map numbers to individual atoms. Unlike atom indices, map numbers remains constant during editing the molecule. Mapping can be useful if you want to identify corresponding atoms in the reactant and product side of a reaction. Map numbers can be added to each atom manually in the Atom properties dialog.
S-groups or substructure groups represent an integral part of a chemical structure. Marvin JS can handle generic S-groups and predefined abbreviated groups (superatom S-groups). Structures containing S-groups can be imported and exported either in MRV or in MDL Molfile format.
Generic S-groups usually represent whole molecules, e.g., solvents. They are visualized between brackets, which will disappear if you remove the group status from the substructure either by using the Ungroup option of the Group pop-up menu or by editing the molecule (changing/adding/deleting atoms or bonds, modifying atom properties...).
An abbreviated group can be part of a larger molecule by connecting the group to other fragments through its attachment point. Currently, only a predefined list of abbreviated groups is available in Marvin JS. Using the abbreviations of chemical functional groups and molecules enables us to create more compact structures. Alternatively, abbreviated groups can be displayed in their full, expanded forms in favor of a more detailed description of the crucial parts of the molecule.
Marvin JS offers a wide selection of abbreviated groups belonging to several chemical compound families (carbohydrates, amino acids, etc.s). These structures are available under the Abbreviated Groups button of the Tools toolbar. Selecting a group in the opening dialog, you can decide whether to use the group in its contracted or expanded form. For the latter option, tick the checkbox in the dialog window. Pressing the "OK" button closes the dialog and puts the selected group on the tip of the cursor, from where you can place it on the canvas with a left-click.
An abbreviated group can be expanded, contracted, or ungrouped after it has been placed on the canvas. These options are available from the Group pop-up menu
2D cleaning of molecules can be performed using the Clean button of the General toolbar. It calculates new 2D coordinates for the atoms on the canvas in order to get the same molecule in an evenly organized fashion.
Partial cleaning of a structure is possible, too. If you have selected fragments of molecules or certain elements of a multi-molecular structure, 2D cleaning affects only these selected objects.
The 2D cleaning feature is available only if the server connection is active, and the appropriate webservice is available, otherwise, the Clean button will not appear on the toolbar.
You can determine the 3D structure of your molecules ("clean them in 3D") by using the Clean 3D on the General toolbar.
The 3D cleaning feature is available only if the server connection is active, and the appropriate webservice is available, otherwise the Clean 3D button will not appear on the toolbar.
Marvin JS only highlights valence errors, but does not correct them, so you should make the necessary corrections manually. In case of recognizing a valence error, Marvin JS highlights the particular atom with red background. This highlight can be turned on and off on the View settings dialog.
Both optical isomerism and geometric isomers can be determined in Marvin JS. In order to assign the R or S label to a chiral center and the E or Z label to a double bond, the Calculate stereo button of the General toolbar should be used. The R/S and E/Z labels are determined via webservice.
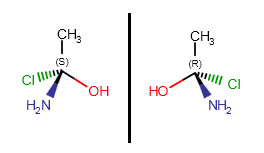
Optical isomers of a molecule differ in the configuration of their chiral atoms (e.g., a carbon atom with four different substituents). Absolute configuration of a chiral center can be marked by R or S stereo labels. If a carbon atom has four different substituents (or three different substituents and an implicit hydrogen) and at least one of them connects to the chiral carbon with a wedge bond, the R or S label can be calculated using the Cahn-Ingold-Prelog priority rules (CIP convention).
Molecules with one chiral center have two optical isomers (enantiomers) which are the mirror images of each other, but they are not superposable.
Enantiomers differ in the absolute configuration of each of their chiral centers.
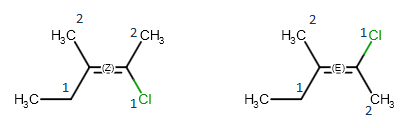
The reason behind geometric isomerism of alkenes is that rotation around double bonds is hindered, so the position of the double bond substituents is fixed relative to each other.
When both double bonded carbon atoms have two different substituents, we can distinguish two non-identical geometric isomers, which are denoted by E and Z labels, respectively.
These stereo labels are calculated by determining the priority of the four substituents using the CIP convention. If the higher ranked substituents are on the same side of the double,
the molecule is the Z isomers, while the E isomer has the higher ranked groups on opposite sides of the double bond.
Query structures are generalized structures representing a group of similar molecules which differ only in some structural elements. For example, all of the molecules possess the same skeleton with a halogen atom as substituent, but it is not crucial to know which halogen exactly. See more about query structures in JChem Query Guide. Using query atoms and bonds offered in Marvin JS different query structures can be designed. Note that atom properties (e.g., atomic charge) can be modified for the query atoms, too. The query atoms are available on the Atoms toolbar, under a "combo" button.
You can define pseudo atoms in your molecules. Replacing an atom with a pseudo will change it to an "Any" type query atom. Some atom properties can also be associated with a pseudo atom on the Atom properties dialog.
The "Absolute" chiral flag on a molecule indicates that every chiral center marked with wedge bonds has a known absolute configuration, that is to say, the structure represents a single, well-defined stereoisomer. This chiral flag can be added to a molecule through the pop-up menu and its display can be turned on/off in the View settings dialog window.
Depending on the file format, a molecule which has wedge bonds without a chiral flag has either of the following meanings:
Marvin JS supports the MDL enhanced stereo representation. Enhanced stereo specifications enable us to refer to several stereoisomers of a molecule by drawing only one structure. Attaching these notations to chiral centers makes it possible to represent stereochemical information: whether we know the absolute configuration of a stereogenic center or we have information only about the relative configuration of two or more chiral atoms. The advantage of enhanced stereo representation is the most palpable when the molecule contains several chiral centers. Illustrating all or even just a few of the molecules stereoisomers can be really demanding in a case like this, but with the help of enhanced stereo specifications only one molecular structure can be enough to represent every isomer.
Marvin JS applies four identifiers, which help us to create groups from the stereocenters. Each group label consists of an identifier, and – in some cases – of a number.
Each and every stereocenter belongs only to one stereogenic group. The following identifiers can be attached to stereogenic centers:
You can find a more detailed description of enhanced stereo representation along with a set of examples in the JChem Query Guide
Please note that import and export of enhanced stereo notations are not possible in MDL Molfile V2000 format, only in the extended MDL Molfile (V3000) format (provided that the relevant webservice is available).
You can add an "Atom List" query atom to your molecule, which covers a set of atoms. The created structure produces a hit if any of the atoms in the list is found in the target. Similarly, you can use "NOT List" query atoms, too. A NOT List query atom can represent any atom that is not part of the list.
Atom Lists and NOT Lists can be defined using the Periodic Table. Pressing the appropriate button on the dialog then clicking on the atoms one after the other prepares the list. Close the Periodic Table when you are ready: the list of atoms is on the cursor. Furthermore, you can replace an atom with a new list or modify an already existing list in the Atom properties dialog, by selecting 'List/NOT list' at the top of the dialog.
A query with an R-group represents substitution variation on the same root structure. An R-group query can involve several derivatives which differ in one or more substituents.
Follow the steps below to create an R-group query with Marvin JS. You can also watch a video tutorial here.
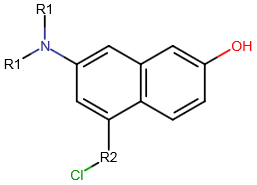
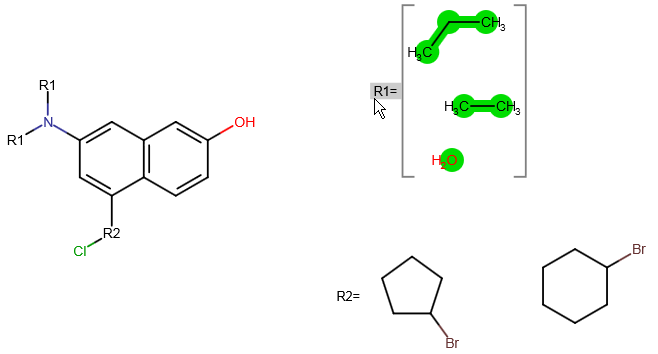
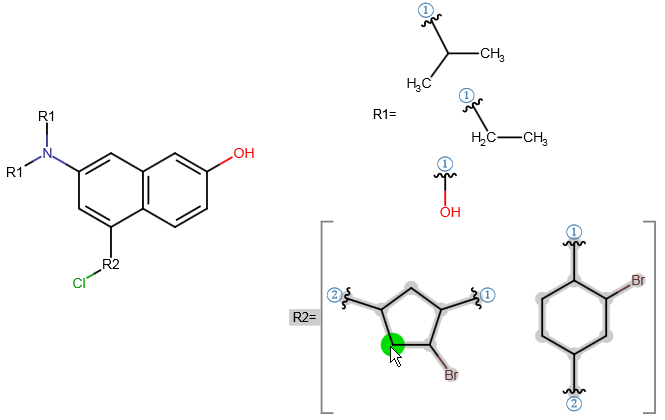
You can edit your R-group definitions by adding fragments to or removing fragments from them. In order to remove a fragment from a group, select this fragment, right-click on the R-label of the group, and select "Remove from this group". If you want to add one or more fragments to an R-group definition, select these fragments, right-click on the R-group label, and choose the "Add to this group" option.
If you do not wish to have an R-group definition any longer, you should right-click on the R-group label, and use the "Ungroup" option in the pop-up menu. This way, the fragments themselves will not be erased, but they will not belong to an R-group definition any longer.
Please note that, for the time being, R-group definition import and export are possible only in ChemAxon Marvin Document (MRV) format or in MDL Extended Molfile (V3000) format provided that the relevant webservice is available.
Built-in homology groups of Marvin JS represent series of chemical compounds with the same specific structural properties. Among these groups there are homologous series groups such as alkyl, alkenyl, alkynyl, carboalicyclyl and other generalized chemical names, like alkali and transition metal etc. Please, follow this link for the complete list of built-in homology groups as well as their aliases (shortened names).
Homology groups are available from the Atom properties dialog after a right-click on an atom. At the top of the dialog, change to "Homology Groups" instead of "Element" then select the appropriate group from the list and choose either its full name or its alias at the "Display" option.