|
DNA, the basic genetic material, is one of the most enigmatic biomolecules and efforts to understand the subtleties of its behaviour from a structural and energetic perspective have not yet been fully rewarded. The pivotal role played by DNA in the synthesis of proteins (gene expression) as well as its own replication makes it an extremely important potential target for drugs , especially for anticancer, antibiotic and antiviral action. Regions of DNA involved in vital processes like origin of replication, promotion of transcription etc. are of particular interest as targets for such drugs.
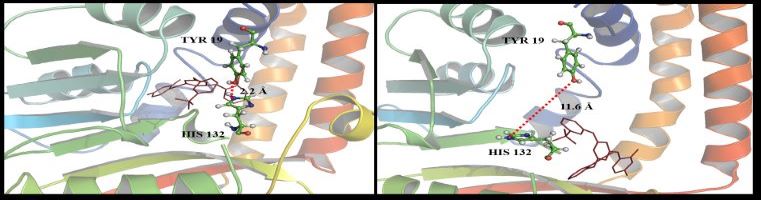
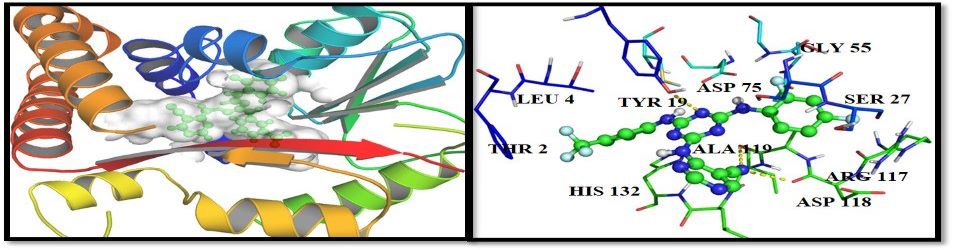
DNA recognition involves a complex interplay of a variety of interactive forces and their consequent energetic contributions - hydrogen bonding interactions, strong electrostatic interactions arising due to phosphates, mobile counter-ions and hydration, van der Waals contacts, the energetic consequences of sequence dependent structural/conformational adaptation of DNA and hydrophobic interactions play critical roles in constructing the energy profile of the binding process. A detailed enumeration and quantification of these energetic contributions can help in addressing vital issues involved in predicting ligand binding free energies and developing a viable methodology for designing or modifying DNA binding small molecules with structural and chemical features that would allow them to recognize the properties of DNA sequences and bind to them with requisite sequence specificity and binding affinity, such that a competition with regulatory proteins could ensue.
Developing a molecular view of the thermodynamics of DNA recognition is essential to the design of ligands for regulating gene expression. Rules governing DNA recognition continue to be elusive. We are endeavoring to tackle this challenge from a standpoint of theory and are in the process of developing and employing a novel energy component strategy which makes it possible to study the molecular thermodynamics of DNA-drug binding make correlations between structure and energetics, thus orienting the study towards eliciting these rules. The computational pathway being developed at our lab employs the MMGBSA methodology wherein molecular mechanical calculations, mainly molecular dynamics simulations, are coupled with the modified generalized Born solvent accessibility model via software developed in-house, and we have been applying it to carry out a systematic characterization of the energetic and structural features that facilitate non-covalent binding of small molecules to DNA.
1.) Shaikh, S.A., Ahmed, S.R. and Jayaram, B. A molecular thermodynamic view of DNA-drug interaction: A case study of 25 minor groove binders. Arch. Biochem. Biophys. 2004, 429, 81-99. [ABSTRACT]
2.)Shaikh S. A. and Jayaram B., A Swift All-Atom Energy Based Computational Protocol to Predict DNA ligand Binding Affinity and ΔTm, J. Med.Chem. , 2007.[ABSTRACT]