|
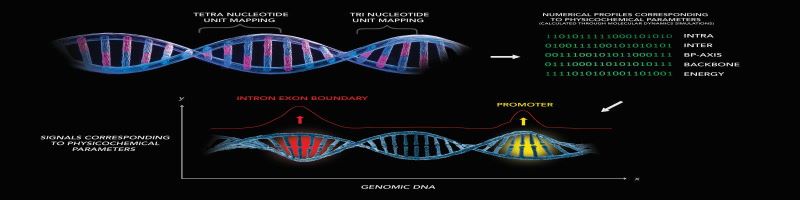
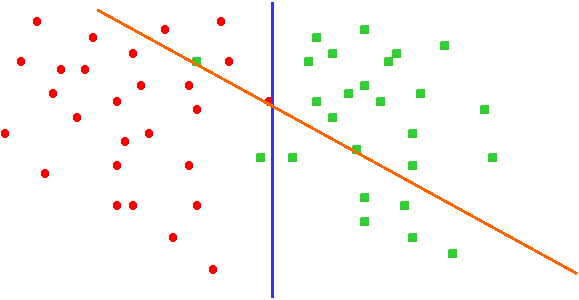
Chemgenome is based on the hypothesis that both the structure of the DNA and its interactions with regulatory proteins and polymerases decide the function of a DNA sequence. It uses a simple three-parameter model based on Watson- Crick hydrogen-bonding energy, base-pair stacking energy, and a third parameter which is related to Protein-Nucleic Acid interactions. Each of these parameters acts as a dimension for a three-dimensional unit vector, whose orientation differs for each trinucleotide. Imagine if you were to separate coding DNA from non-coding DNA. Lets imagine that we found a way to represent each DNA sequence as a dot on a 2D plane. All the coding DNA sequences are represented as Red dots while the non-coding DNA sequences are represented as Green dots, as below.

We can separate the two clusters of dots using a line. In Fig 2, we can see two separating lines, but evidently the blue line separates more effectively than the green line.
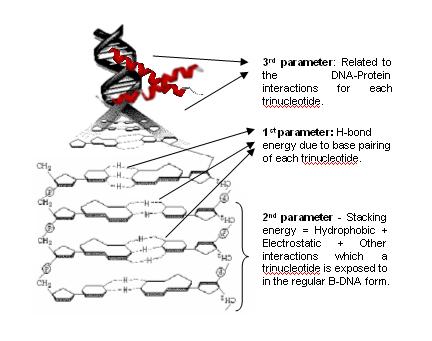
▪The first component was constructed by finding the Hydrogen bonding energy of Trinucleotides(codons).
▪The second component was constructed by finding the Stacking energy(sum of electrostatic, hydrophobic and other forces which the trinucleotides are exposed to when it is stacked with other nucleotides in the B-DNA form).
▪The third component of the vector reflects the DNA protein interactions. It's assignment follows the “Conjugate” rule which is derived from the Wobble Hypothesis (For more on conjugate rule, refer to [Jayaram, B. Beyond the wobble: the rule of conjugates. J. Mol. Evol. 1997, 45, 704-705].
S.No. |
NCBI_ID |
Species Name |
Genes |
TP# |
FP# |
SS# |
SP# |
CC# |
1 |
NC_000117 |
Chlamydia trachomatis |
463 |
458 |
4 |
0.98 |
0.99 |
0.98 |
2 |
NC_000853 |
Thermotoga maritima MSB8 |
641 |
619 |
3 |
0.96 |
0.99 |
0.96 |
3 |
NC_000854 |
Aeropyrum pernix K1 |
561 |
532 |
7 |
0.94 |
0.98 |
0.93 |
4 |
NC_000868 |
Pyrococcus abyssi GE5 |
632 |
630 |
241 |
0.99 |
0.63 |
0.49 |
5 |
NC_000907 |
Haemophilus influenzae |
955 |
953 |
7 |
0.99 |
0.99 |
0.99 |
6 |
NC_000908 |
Mycoplasma genitalium G-37 |
189 |
186 |
2 |
0.98 |
0.98 |
0.97 |
7 |
NC_000909 |
Methanocaldococcus janaschii |
720 |
708 |
9 |
0.98 |
0.98 |
0.97 |
8 |
NC_000912 |
Mycoplasma pneumoniae M129 |
243 |
241 |
2 |
0.99 |
0.99 |
0.98 |
9 |
NC_000913 |
Escherichia coli K12 |
2759 |
175 |
659 |
0.63 |
0.72 |
0.39 |
10 |
NC_000915 |
Helicobacter pylori |
731 |
727 |
4 |
0.99 |
0.99 |
0.98 |
11 |
NC_000916 |
Methanobacterium thermoautotrophicum |
719 |
711 |
4 |
0.98 |
0.99 |
0.98 |
12 |
NC_000917 |
Archaeoglobus fulgidus |
782 |
774 |
8 |
0.98 |
0.98 |
0.97 |
13 |
NC_000917 |
Archaeoglobus fulgidus DSM4304 |
782 |
774 |
8 |
0.98 |
0.98 |
0.98 |
14 |
NC_000918 |
Aquifex aeolicus VF5 |
584 |
575 |
3 |
0.98 |
0.99 |
0.97 |
15 |
NC_000921 |
Helicobacter pylori strain J99 |
658 |
648 |
9 |
0.98 |
0.98 |
0.97 |
16 |
NC_000922 |
Chlamydophila pneumoniae CWL029 |
597 |
590 |
9 |
0.98 |
0.98 |
0.97 |
17 |
NC_000948 |
Borrelia burgdorferi B31 plsmids cp32-1 |
11 |
11 |
0 |
1.0 |
1.0 |
1.0 |
18 |
NC_000949 |
Borrelia burgdorferi B31 plsmids cp32-3 |
11 |
11 |
0 |
1.0 |
1.0 |
1.0 |
19 |
NC_000950 |
Borrelia burgdorferi B31 plsmids cp32-4 |
11 |
11 |
0 |
1.0 |
1.0 |
1.0 |
20 |
NC_000951 |
Borrelia burgdorferi B31 plsmids cp32-6 |
10 |
10 |
0 |
1.0 |
1.0 |
1.0 |
Chemgenome 2.0 goes a step further and predicts the coding regions in Prokaryotes if a whole genome or part of genome is given as an input.
To know more about this physico-chemical model, refer to
[Dutta, S., Singhal, P., Agrawal, P., Tomer, R., Kritee, Khurana, E. and Jayaram, B. A Physico-Chemical Model for Analyzing DNA sequences. J. Chem. Inf. Model., 2006, 46(1), 78-85. ] ABSTRACT