|
Pursuing the dream that once the gene target is identified & validated drug discovery protocols could be automated using Bioinformatics & Computational Biology tools, our research group at IITD has developed an active site directed drug design protocol and software. The suite of programs (christened �Sanjeevini�) has the potential to evaluate and /or generate lead-like molecules for any biological target. |  |
| Sanjeevini Pathway | |
The various modules of this suite are designed to ensure reliability and generality. The software is currently being optimized on Linux and Sun clusters for faster and better results. The methodology consists of design of a library of templates, generation of candidate inhibitors, screening candidiates via drug-like filters, parameter derivation via Quantum Mechanical calculations for energy evaluations, Monte Carlo Docking and binding affinity estimates based on post facto analyses of all atom molecular dynamics trajectories. The protocols tested on Cyclooxygenase-2 as a target could sucessfully distinguish NSAIDs (Nonsteroidal Anti-inflammatory drugs) from non-drugs. Validation on other targets is in progress. |
 |
|---|---|
| Result for Cox2 An energy separation between drugs and non drugs |
|
 |
The binding free energies of COX-2 with NSAIDs (blue) versus non-drugs (red) are shown in the figure. A clear separation of NSAIDs from non-drugs is discernible. Overall, drugs (NSAIDs) which are supposed to bind to the target yield negative binding free energies and non-drugs, i.e., molecules which are not supposed to bind yield positive binding free energies. This is a redeeming feature and allays any apprehensions that the lead predicted may not bind in vitro and vice versa. A related concern that any molecule can be positioned in the active site of the target and energy minimized but may not work as a drug is addressed by focusing on free energy methodologies which are compute intensive but thermodynamically sound and reliable. Automated sorting of drugs from non-drugs seems possible. |
DNA plays a pivotal role in the synthesis of proteins (gene expression) as well as its own replication making it an extremely important potential target for drugs, especially for anticancer, antibiotic and antiviral action.
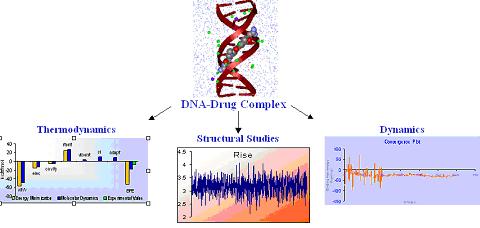
DNA recognition involves a complex interplay of a variety of interactive forces. Hydrogen bonding interactions, strong electrostatic interactions arising due to phosphates, mobile counter-ions and hydration, van der Waals contacts, the energetic consequences of sequence dependent structural/conformational adaptation of DNA and hydrophobic interactions play critical roles in constructing the energy profile of the binding process. Thus, developing a molecular view of the thermodynamics of DNA recognition is essential to the design of DNA binders.
We are endeavoring to tackle this challenge from a standpoint of theory. Currently we are carrying out studies on the structure, dynamics and thermodynamics of DNA-drug complexes. Our computational strategy involves free energy component analyses which enables the study of molecular thermodynamics of DNA-drug binding and make correlations between structure and energetics, thus orienting the study towards eliciting pointers for drug design.
REFERENCE :
1.) Jayaram, B., Latha, N.,Jain, T., Sharma, P., Gandhimathi, A., Pandey, V.S., Sanjeevini: A Comprehensive Active-Site Directed Lead Design Software. Indian Journal of Chemistry-A. 2006,
45A, 1834-1837.
(in press)
2.) Jain, T. and Jayaram, B. An all atom energy based computational protocol for predicting binding affinities of protein-ligand complexes, FEBS Letters, 2005, 579, 6659-6666.[ ABSTRACT ]
3.) Latha, N., and Jayaram, B. A Binding Affinity Based Computational Pathway for Active-Site Directed Lead Molecule Design: Some Promises and Perspectives. Drug Design Reviews-Online. 2005, Vol2(2), 145-165. [ABSTRACT]
4.) Shaikh, S.A., Ahmed, S.R. and Jayaram, B. A molecular thermodynamic view of DNA-drug interaction: A case study of 25 minor groove binders. Arch. Biochem. Biophys. 2004, 429, 81-99. [ABSTRACT]
5.) Latha, N., Jain, T., Sharma, P. and Jayaram, B. A free energy based computational pathway from chemical templates to lead compounds: a case study of COX-2 inhibitors. J. Biomol. Struct. Dyn. 2004, 21, 791-804.[ABSTRACT]
6.) Kalra, P., Reddy, T.V. and Jayaram, B. Free energy component analysis for drug design: a case study of HIV-1 protease-inhibitor binding. J. Med. Chem. 2001, 44, 4325-4338. [ABSTRACT ]