A genome is an organism.s complete set of DNA, comprising of nuclear and mitochondrial DNA. Each genome contains all of the information needed to build and maintain that organism. A human haploid cell, consist of 23 nuclear chromosome and one mitochondrial chromosome, contains more than 3.2 billion DNA base pairs.
Eukaryotic genome is linear and conforms the Watson-Crick Double Helix structural model. Embedded in Nucleosome-complex DNA & Protein (Histone) structure that pack together to form chromosomes. Eukaryotic genome have unique features of Exon - Intron organization of protein coding genes, representing coding sequence and intervening sequence that represents the functionality of RNA part inside the genome.
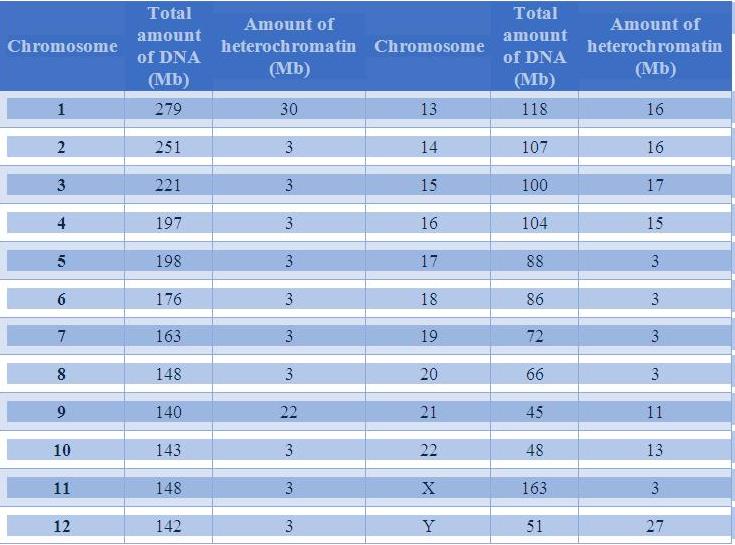
Table 1: DNA content of Human Chromosomes
Configuration of Eukaryotic genome:
The configuration of eukaryotic genome includes protein coding region, gene regulating region, gene related sequence and intergenic DNA or extra genic DNA which includes low copy number and moderate or high copy number repetitive sequence, the flow chart representation of configuration is given below:

1. GENES AND GENE RELATED SEQUENCES
A. PROTEIN CODING REGION (EXONS)
. Protein coding sequences are the DNA sequences that are transcribed into mRNA later translated to proteins.
. The complete protein coding genes capacity of the genome is contained within the exomes (the part of the genome formed by exons, the sequences which when transcribed remain within the mature RNA sequence after introns are removed using RNA splicing)and consists of DNA sequences encoded by exons that can be later translated into proteins. .
. It consists of ORF (Open reading frame). These are the reading frame that has the potential to code for the proteins/peptide. It is stretch of codons that do not contain a stop codon (UAA, UAG, and UGA). An AUG with the ORF may indicate where translation starts.
1. Get all "gene" lines:
awk '{if($3=="gene"){print $0}}' file_name
2. Get all sequence of "gene"
awk '{if($3=="gene"){getline; print $0}}' file_name
3. Get all "protein-coding transcript" lines:
awk '{if($3=="transcript" && $20=="\"protein_coding\";"){print $0}}' file_name
4. Get all the "protein-coding gene" lines:
awk -F "\t" '$3 == "gene" { print $9 }' file_name | \
awk -F "; " '$3 == "gene_type \"protein_coding\""' | \
head | less .S
5. Get all the "protein-coding exon" lines:
awk -F "\t" '$3 == "exon" { print $9 }' file_name | \
awk -F "; " '$3 ~ "protein_coding"' | \
head | less -S
6. Get level 1 & 2 annotation (manually annotated) only:
awk '{if($0~"level (1|2);"){print $0}}' file_name
7. Get the gene type of each gene:
awk -F "\t" '$3 == "gene" { print $9 }' file_name | awk -F "; " '{ print $3 }' | head | less -S
8. Get the number of exon in each gene: (similar for transcript and CDS)
awk -F "\t" '$3 == "exon" { print $9 }' file_name | \ tr -d ";\"" | \ awk -F " " '
$6 == "protein_coding" { gene_counter[$10] += 1}
END { for (gene_name in gene_counter){ print gene_name, gene_counter[gene_name] }
}' > number_of_exons_by_gene.txt
9. Get the start and stop position of all the gene
awk '{if($3=="gene"){print $4"\t"$5}}' file_name
|
