|
Combining bioinformatics tools and ab initio methodologies, considerable progress has been made towards a pathway that is computationally expeditious for tertiary structure prediction of small proteins.
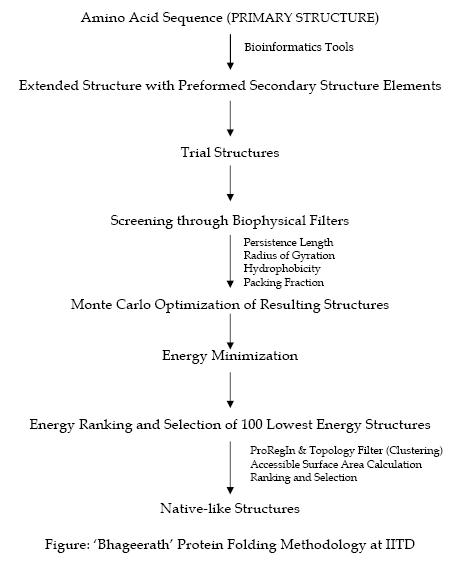
The software suite Bhageerath comprises eight modules configured to function independently or in a conduit. Starting with amino acid sequence(primary structure) and secondary structure information (helix/sheet/loop) of a protein in the first module, multiple three dimensional atomic level structures are generated sampling the conformational space of the loop dihedrals in the second; in the third module a set of biophysical filters (persistence length, radius of gyration etc.) are applied which are designed to screen the trial structures to reduce the sample size. The resultant structures are refined in the fourth module by a Monte Carlo sampling in dihedral space to remove steric clashes / overlaps in 3-D space. An atomic level energy optimization is carried out in the fifth module and the structures scored based on energy in the sixth. Module seven reduces the probable candidates based on the protein regularity index of the(phi) φ and (psi) ψ dihedral values and module eight further reduces the structures selected to 10 using topological equivalence criterion and accessible surface area.
Thus millions of possible structures for a given protein sequence are brought down to 10 candidate structures with the possibility of bracketing the native in these 10. Results on a few small globular helical proteins have shown that native like folds in the root mean square deviation (RMSD) range of 3-6 A are captured in the best 10 structures energy-wise in all the cases without any exception. The ‘needle in a hay stack problem’ is thus reduced to choosing the best candidate from among the 10 lowest energy structures.
Comparison with the existing bioinformatics tools suggests the performance of the present methodology to be satisfactory and useful particularly when the database is deficient in sequence homologues. Preliminary work on alpha/beta systems has yielded encouraging results. Currently the expected prediction time with Bhageerathweb server for systems with two secondary structures (with one loop in between) is ~4-5 min; while for systems with three secondary structures (with two loops in between), it is ~2-3 h on a dedicated 32 processor cluster. Attempts to extend the methodology to larger systems with reduction in computational times are in progress.
1. Jayaram, B., Bhushan, K., Shenoy, S. R., Narang, P., Bose, S., Agrawal, P., Sahu, D., Pandey, V.S. Bhageerath : An Energy Based Web Enabled Computer Software Suite for Limiting the Search Space of Tertiary Structures of Small Globular Proteins. Nucl Acids Res., 2006. , 34, 6195-6204; doi: 10.1093/nar/gkl789 [Full Paper]
2. Narang, P., Bhushan, K., Bose, S., and Jayaram, B. Protein structure evaluation using an all-atom energy based empirical scoring function, J. Biomol.Str.Dyn, 2006 23, 385-406. [ABSTRACT]
3. Narang, P., Bhushan, K., Bose, S. and Jayaram, B. A computational pathway for bracketing native-like structures for small alpha helical globular proteins. Phys. Chem. Chem. Phys., 2005 7, 2364-2375. [Full Paper]
4.Thukral, L., Shenoy, S. R., Bhusan, K and Jayaram, B. ProRegIn: A Regularity Index for the Selection of Native- like Tertiary Structures of Proteins, Journal of Biosciences 2007. 32(1), 71-81. 2007 7, 2364-2375. [Full Paper]